The transfer of genetically modified T cells into patients for the treatment of cancer has demonstrated impressive response rates in some types of leukemia and lymphoma. These observations provide crucial proof-of-concept that adoptive immunotherapy with engineered T cells can be an efficacious strategy. As a result, the academic and biopharmaceutical communities now have the necessary confidence to redouble their efforts in order to extend the reach of this technology to broader cancer types.
Introduction
The tools of modern cancer therapy include surgery, radiation, chemotherapy, oncogenic pathway inhibition, and immunotherapy. The discipline of immunotherapy takes advantage of cellular components of the normal immune system in order to generate or enhance a pre-existing anti-cancer response. The immune system is designed to discriminate self from non-self and uses specialized effector cells to attack non-self entities such as infectious agents and diseased cells. These effector cells include T cells, natural killer (NK) cells, phagocytes such as neutrophils or monocyte/macrophages, and antibody-secreting plasma cells. A theoretical advantage of immunotherapy is that it can be exquisitely specific towards cancer cells without causing toxicity to normal tissues. In contrast, surgery, radiation and chemotherapy are all relatively blunt tools. The specificity of immunotherapy is related to a stereotyped receptor-ligand interaction that is akin to a lock and key mechanism. In this analogy receptors, present on immune effector cells, are the lock and their ligands, present on target cells, are the key. Each receptor can only be activated by its specific ligand. Upon activation of the receptor, multiple functions of the immune effector can be triggered simultaneously.
T cells normally recognize their target using a T cell receptor (TCR). The TCR is activated upon binding a “key” comprised of a peptide nestled within the groove of the major histocompatibility antigen (MHC) molecule . The peptide is typically derived from within the target cell. T cells bearing a TCR that is activated by self-antigens are deleted in the thymus in order to prevent attack of the body’s own tissues (since this would result in autoimmune disease) and therefore all T cells that circulate in the body theoretically have the capacity to recognize foreign antigens such as infectious agents or mutated peptides. While most cancer cells contain at least one DNA mutation (and hence should be recognized as being foreign) not all these mutations can be adequately presented within the MHC and therefore there is not a key for every lock. Furthermore, even where there is an adequate lock-and-key receptor-ligand pair, the strength of interaction between the TCR and its ligand is typically lower for cancer cells than for microbes. This means that many cancer cells can go undetected by the body’s repertoire of T cells, or if they are detected the strength of interaction is insufficient to trigger the degree of T cell activity that would result in successful attack of the cancer cells.
Antibodies are soluble proteins that are produced by plasma cells and can recognize a much greater diversity of antigen types than T cell receptors. These antigens include carbohydrate, peptide, glycolipid, glycopeptides or other domains. Antibodies do not require their antigen to be presented within the MHC molecule and typically have a higher affinity for their ligand than do T cell receptors. Antibody binding to antigen can recruit immune cells or other effector functions such as complement in order to directly kill cancer cells, or alternatively can block or neutralize interactions between cancer cells and various pro-survival signals that they require. Of note, antibodies can only recognize antigens that are present on the outside membranes of target cells, whereas most cancer-associated mutated peptides are located within the cell.
The above observations explain how the immune system should work to prevent cancer from occurring. However, by the time cancer has developed, the immune system has by definition failed to protect against its development. At this point, options to recruit the power of the immune system can be divided into two broad strategies. Firstly, the endogenous immune system can be re-awakened by removing the inhibitory signals that have been co-opted by cancer cells to dampen immune attack (immune checkpoint inhibition) or by stimulating it using cancer vaccines. The second approach is called adoptive cellular therapy, whereby immune cells are first removed from the body of the patient or of a healthy donor, altered or enhanced in some way, and then infused back into the patient.
Genetic engineering uses the tools of molecular biology to manipulate the basic building blocks of biology (nucleic acids) in order to enhance naturally occurring structure or function. Genetic engineering thus leads to the concept of “synthetic biology” – loosely, creating something better than nature. In essence, synthetic biology is directed, accelerated evolution. In the context of adoptive cellular immunotherapy, T cells and other immune cells can be engineered to exhibit features that they do not normally have.
In this essay I will describe advances in genetically engineered cellular therapy. I will focus on T cells and specifically on two ways to re-direct their recognition of cancer antigens: TCR-based and chimeric antigen receptor (CAR)-based (Fig.1).
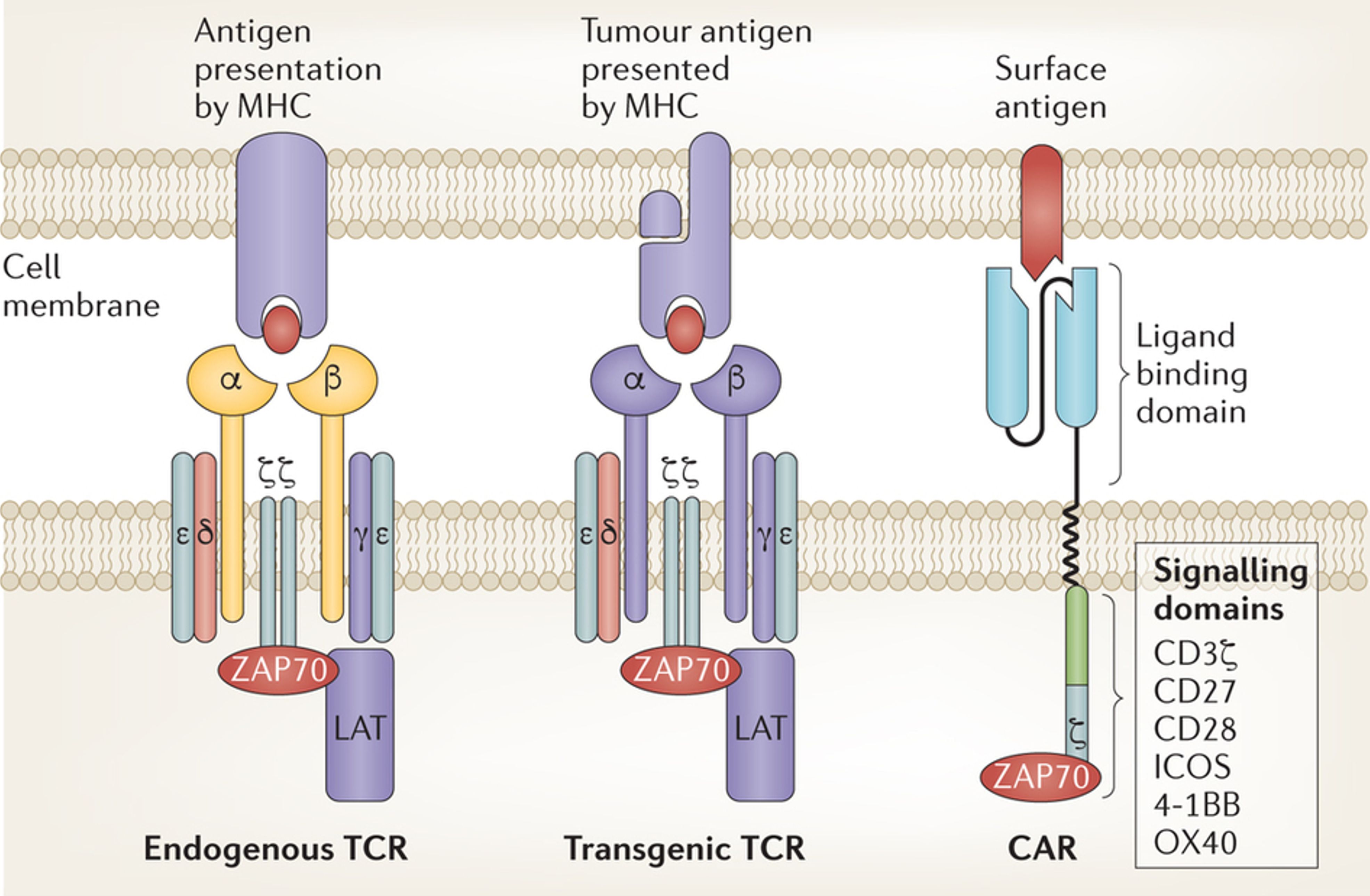
TCR-transgenic T cells
The body’s repertoire of T cells is necessarily very broad and since there is a finite number of T cells that the body can produce and host, the repertoire is not very deep. This means that there are relatively few T cells with any given specificity. As mentioned earlier, additional barriers to the recognition by T cells of cancer cells using their T cell receptors include deletion of auto-reactive T cells and the relatively low affinity of TCR for their target antigens. These three hurdles can be overcome by temporarily removing a large number of T cells from the body and instructing them to produce TCR of known high specificity and affinity. These T cells are known as transgenic TCR-T cells. If the genetic identity (nucleotide coding sequence) of a TCR toward a cancer antigen of interest is known, that nucleotide sequence can be inserted into the T cells in the laboratory using a vector (typically, a virus that is specialized at infecting T cells and transferring its DNA into them). The nucleotide sequence can be designed so that the resultant lock-and-key interaction is very tight, thereby increasing the affinity of interaction, and these are known as affinity-enhanced TCR. However, the introduced genetically engineered TCR can sometimes interact with the original TCR of each of the T cells and very occasionally this creates a hybrid TCR with unintended specificity and the potential to attack the body’s normal tissues. Additional unintended specificities can arise when affinity enhancement of the TCR leads to a “sticky” lock-and-key interaction such that the engineered T cells bind and attack tissues bearing a “lock” (or ligand) that is similar yet not identical to their intended target. Nonetheless, clinical trials have been conducted in patients with melanoma, myeloma, and sarcoma using TCR-transgenic T cells and have produced clinical responses . Future directions include enhancing the function of the TCR-transgenic T cells, and finding new TCR for cancer-associated antigens. However, given the TCR recognize peptides within the MHC groove as described above, this strategy is always going to be limited to a certain category of antigens (derived from proteins) and will fail when cancer cells eliminate the MHC molecule from their surface. Unfortunately the latter is an all-too-common escape strategy used by cancer cells.
CAR T cells
The mythical beast Chimaera was composed of several different animals. Similarly, Chimeric Antigen Receptor (CAR) T cells are derived from components of several different immune cells. Most are T cells that are instructed to produce a synthetic antigen receptor consisting of the ligand-recognizing portions of an antibody which is fused to a selection of intracellular signalling domains for T cell activation. This confers on the genetically engineered T cells, for the first time, the ability to recognize a wide variety of antigens on the surface of cancer cells in a non-MHC restricted manner . First-generation CAR engineering includes the receptor only. Second- and subsequent generation CAR engineering includes additional features that further enhance stimulation of the T cells upon recognizing their antigen. Again genetic engineering occurs outside the body in the laboratory and typically uses a viral vector to deliver the engineered genetic material into the T cells. Another aspect of the manufacturing process of the T cells in the laboratory is that the cells can multiply several-fold (typically undergoing approximately five doublings to expand 25-fold) thus generating a T cell product that is significantly more numerous than at the beginning of the manufacturing process. At the conclusion of this process, that normally takes approximately two weeks, the T cells can be administered to the patient via an intravenous infusion that takes a few minutes, called adoptive transfer.
Upon intravenous infusion the T cells can theoretically migrate to all tissues and organs, although they are probably better suited to home to blood forming organs and components of the immune system such as lymph nodes. T cells are able to penetrate large masses of cancer although they do find it difficult to thrive within those masses, due to a variety of factors that include physical barriers, chemically unfavorable environments and active evasion strategies exerted by the cancer. Within the tissue, T cells crawl around until they encounter their antigen. They then engage with the target cells that bear the antigen, and are activated through their receptor (Fig.2). Activation leads to the production of killing molecules and to self-replication of the T cells to produce daughter cells that are capable of more killing, and that upon the eradication of cancer persist as memory cells in order to prevent future relapses.
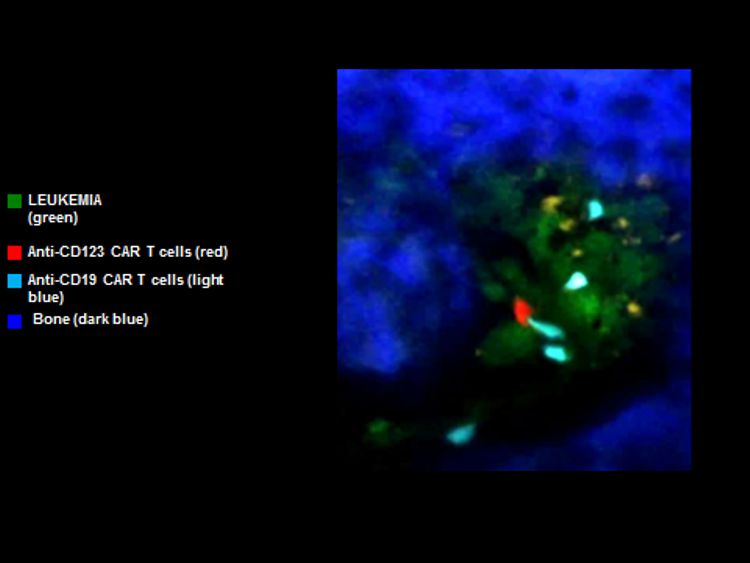
To date the best results with CAR T cells have been achieved in blood cancers such as leukemia. Indeed, the majority of patients with very advanced B-cell acute lymphoid leukemia (B-cell ALL) that resisted multiple prior attempts at treatment achieve complete elimination of their disease . Complete responses have also been achieved in many patients with another blood cancer known as lymphoma . In contrast, attempts to harness the CAR T cell platform for the treatment of solid cancers such as bowel, breast, ovarian or kidney have been considerably less successful. While this discrepancy relates in part to the difficulty that T cells experience in penetrating solid tumor masses, it is likely that even if this hurdle were overcome, there would be an even higher bar to clear: Since there are virtually no tumor-specific antigens on the surface of cancer cells, powerful CAR T cell approaches directed against antigens that are shared by cancerous tissues and their normal counterparts are just as likely to attack the normal tissues. Indeed, this is already the case with the most successful CAR T cell therapies that are directed against B-cell malignancies such as ALL: both malignant and normal B cells are eradicated. However humans can tolerate prolonged periods without normal B cells. In contrast, it is likely that CAR T cell attack of normal tissues such as liver or bowel would cause unacceptable side effects.
Summary and future prospects
To date, adoptive transfer of genetically engineered T cells has been performed in a variety of blood cancers and solid tumors. The most exciting results have been obtained in patients with blood cancers since (i) they are easily accessed by CAR T cells and (ii) their normal counterparts are to some extent dispensable (or their function can be easily replaced by supportive therapies). The two major impediments that currently prevent progress with genetically engineered T cell therapies are: (i) the need for tumor-specific antigens that are not shared with normal cells, and (ii) mechanical, chemical and immunologic barriers to the penetration of large tumor masses by T cells. The successful treatment of B-cell ALL using CAR T cells and the promising results in cancers such as multiple myeloma and melanoma using TCR-transgenic T cells have generated a groundswell of enthusiasm on the part of scientists and pharmaceutical companies. This enthusiasm will undoubtedly lead to the stepwise resolution of the barriers that currently prevent more widespread application of genetically engineered T cell therapy for cancer.
Further reading on this topic:
- Fesnak AD, June CH, and Levine BL (2016) Engineered T cells: the promise and challenges of cancer immunotherapy Nat Rev Cancer 16: 566-581
- van der Stegen SJC, Hamieh M, and Sadelain M (2015) The pharmacology of second-generation chimeric antigen receptors Nat Rev Drug Discov 14: 499-509
- Kalos M and June CH (2013) Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology Immunity 39: 49-60
Other Articles by This Author
Further Articles in  Medical Research
Medical Research
 This work is distributed under a Creative Commons Attribution ShareAlike 4.0 International License (CC BY-SA 4.0). Its use, distribution and reproduction in other forums is permitted provided both the original author(s) and Capeia are accredited, and the license is unchanged (CC BY-SA 4.0). Images or other third party material in this article are covered under the article’s Creative Commons license unless otherwise indicated; if the material is not covered under a Creative Commons license, users will need to obtain permission from the license holder before it can be reproduced.
This work is distributed under a Creative Commons Attribution ShareAlike 4.0 International License (CC BY-SA 4.0). Its use, distribution and reproduction in other forums is permitted provided both the original author(s) and Capeia are accredited, and the license is unchanged (CC BY-SA 4.0). Images or other third party material in this article are covered under the article’s Creative Commons license unless otherwise indicated; if the material is not covered under a Creative Commons license, users will need to obtain permission from the license holder before it can be reproduced.
Responses